Welcome to the Tutorial!
DEGRONOPEDIA is an online resource that allows inspection and visualization of known degron motifs in the proteomes of selected model organisms, as well as in a user-submitted sequence or structure, and many more.
Discover its features in our video ⚡quick tour below! 🎬
Few words of introduction
In the ubiquitin-proteasome system (UPS), E3 ubiquitin ligases are critical for tagging proteins for degradation, primarily attaching ubiquitin to their lysine residues, with emerging evidence indicating cysteine, serine, and threonine residues also acting as ubiquitination sites. Proteins are targeted for degradation through degrons, typically short linear motifs found in disordered regions, which can be constitutive or conditional, the latter emerging from post-translational modifications. Degrons are not restricted in location but those at the amino or carboxyl termini, integral to the N- or C-degron pathways, have been extensively studied.
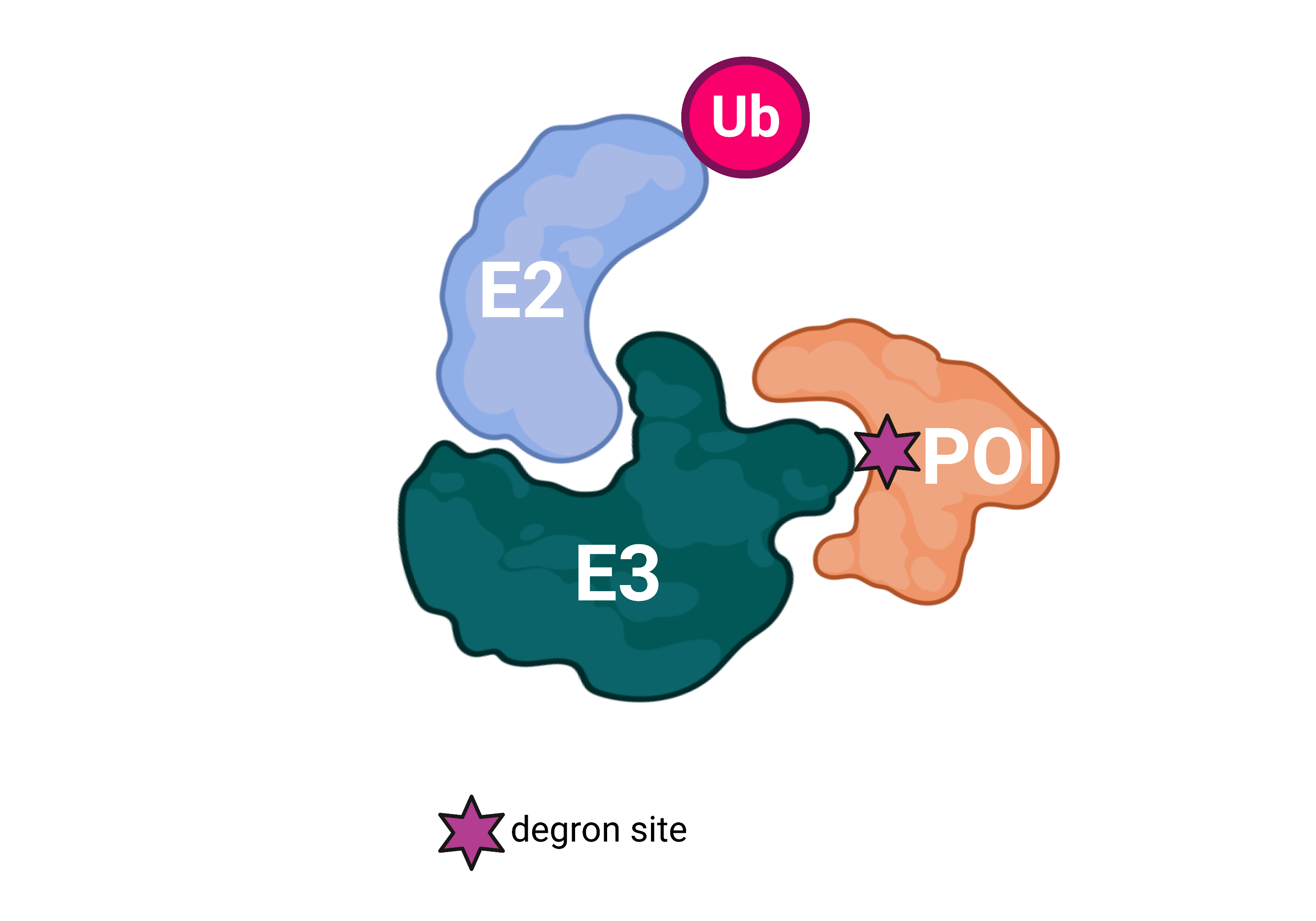
E2 - ubiquitin-conjugating enzyme, E3 - ubiquitin ligase, Ub - ubiquitin, POI - protein of interest.
Recent research suggests that ubiquitination and subsequent degradation depend not just on degron recognition. The tripartite degron model proposed by Guharoy et al. introduces secondary degrons, comprising lysine residues near the primary degron, and tertiary degrons, which are flexible, intrinsically disordered regions (IDR) nearby, facilitating protein unfolding before proteasomal entry. The secondary and tertiary degrons are suggested to play subsidiary roles that affect ubiquitin-signaling. Deficiency of one of the elements of the tripartite degron model, e.g., an IDR near a ubiquitinated lysine, can result in non-proteolytic ubiquitination functions.
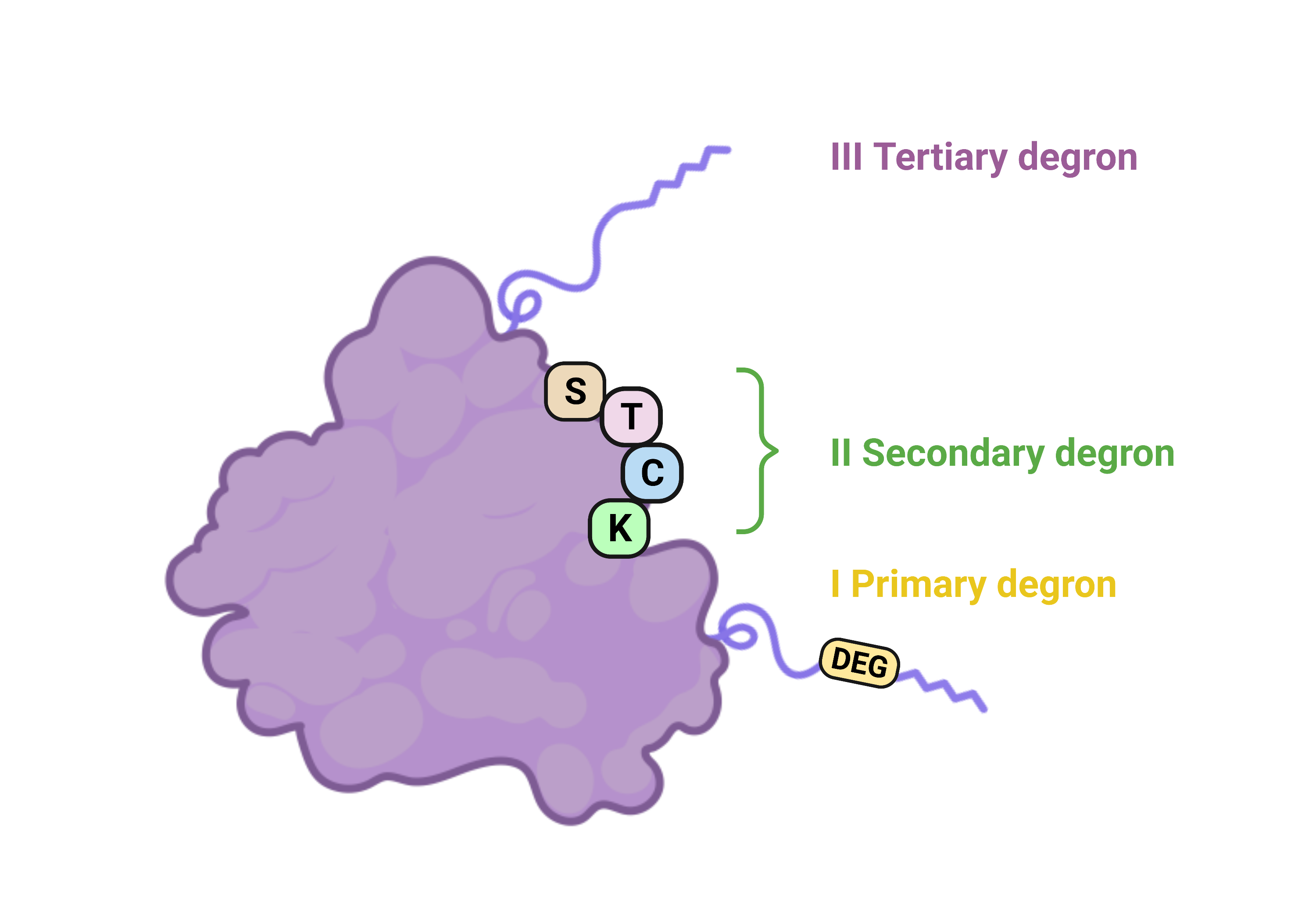
E2 - ubiquitin-conjugating enzyme, E3 - ubiquitin ligase, Ub - ubiquitin, POI - protein of interest.
Addressing these complexities, we introduce DEGRONOPEDIA, a web server for identifying and analyzing degron motifs in proteins.
It provides:
- degron motifs occurring in the query
- structural context (solvent-accessibility, intrinsically disordered region, secondary structure) for each degron motif found
- post-translational modifications and mutations occurring within each degron motif and its flanking regions
- comprehensive visualization of the location of degron motifs in the sequence with structural data, post-translational modifications and mutations
- evolutionary conservation of degron motifs among orthologous proteins
- degron context according to the tripartite degron model1
- Gravy hydrophobicity index for N-/C-terminus of the query
- experimental or Machine Learning-predicted Protein Stability Index (PSI) of N-/C-terminus of the query (provides insight into protein termini stability)
- simulations of proteolytic cleavage followed by screening for degron motifs in each newly emerged N-/C-termini after proteolysis
- E3 ubiquitin ligases interacting with the query
⚡Quick Tour
 Watch tutorial on YouTube
Watch tutorial on YouTube